A Scientific Article by Ms. Zainab Mahmoud Hatem Titled: Thalassemia Disease
Thalassemia Disease: A Comprehensive Scientific Overview
Abstract
Thalassemia is a group of inherited blood disorders that affect hemoglobin synthesis, leading to chronic anemia with severity ranging from mild cases to severe, life-threatening conditions. Thalassemia is more prevalent in the Mediterranean region, the Middle East, and South Asia. This article aims to review the genetic, pathological, diagnostic, and therapeutic aspects of thalassemia, with a focus on the role of medical laboratories in diagnosis and follow-up.
Introduction
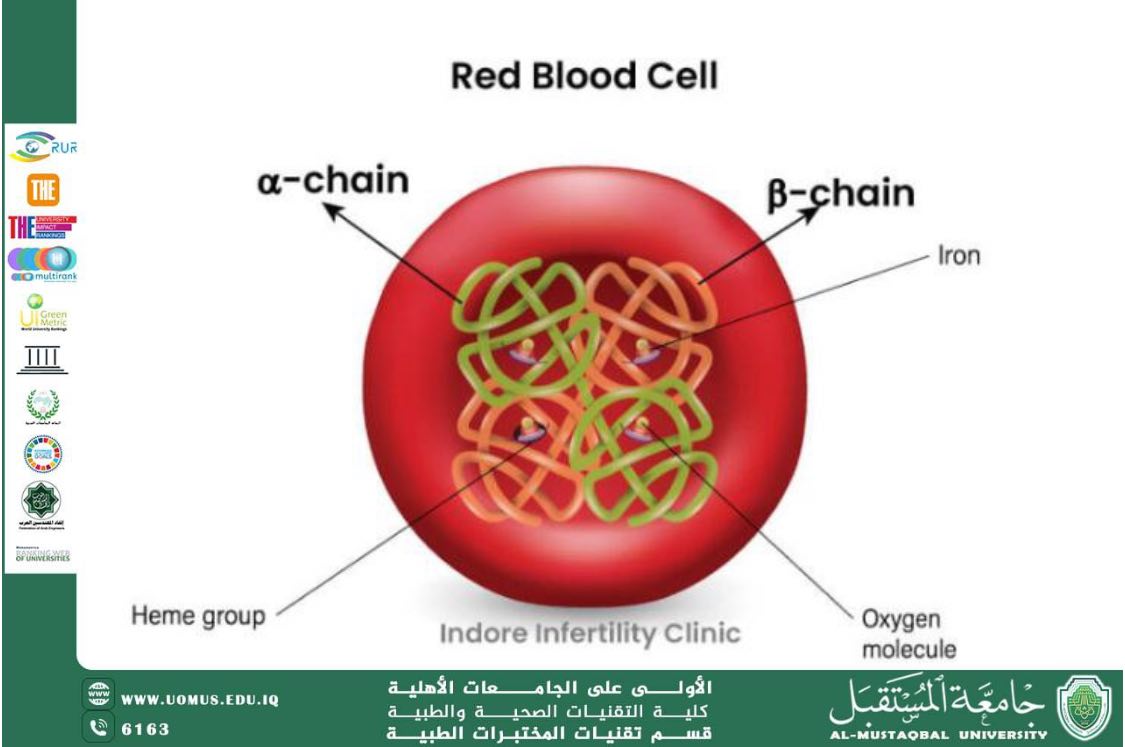
Hemoglobin is the main component of red blood cells and is responsible for transporting oxygen to body tissues. Any defect in its structure or production leads to blood disorders, among which thalassemia is one of the most significant. The disease results from genetic mutations affecting globin chain synthesis, leading to a shortened lifespan of red blood cells and the development of hemolytic anemia.
Classification and Types of Thalassemia
Thalassemia is classified into two main types depending on the affected hemoglobin chain:
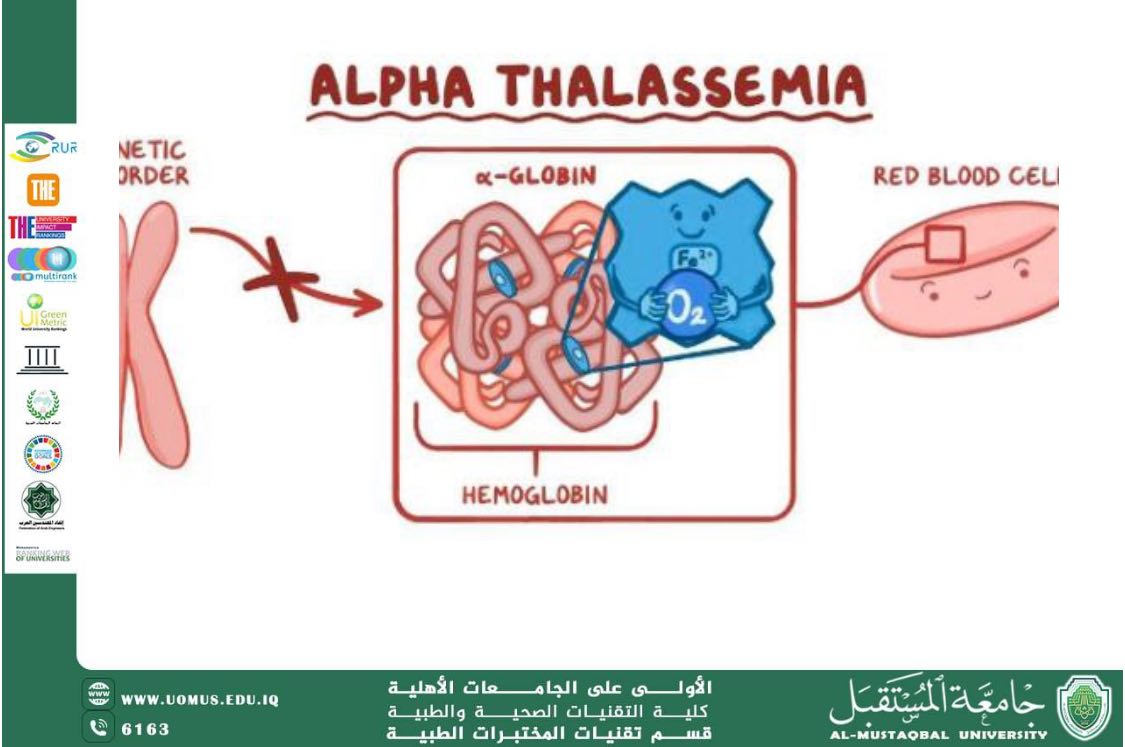
Alpha Thalassemia (α-Thalassemia)
This type results from defects in the genes responsible for alpha-globin chain production. Its severity ranges from silent carriers to severe conditions such as hydrops fetalis.
Beta Thalassemia (β-Thalassemia)
This type results from reduced or absent synthesis of beta-globin chains and is classified into:
• Beta Thalassemia Minor
• Beta Thalassemia Intermedia
• Beta Thalassemia Major
Pathophysiology
Defective globin chain synthesis leads to an imbalance between globin chains, causing precipitation of abnormal chains inside red blood cells. This results in premature destruction of red blood cells in the bone marrow and spleen, leading to chronic anemia, splenomegaly, and bone deformities due to bone marrow hyperactivity.
Clinical Manifestations
Clinical symptoms vary depending on the type and severity of the disease and include:
• Pallor and chronic fatigue
• Growth retardation in children
• Enlargement of the spleen and liver
• Facial and skull bone deformities
• Iron overload due to repeated blood transfusions
Laboratory Diagnosis
Medical laboratories play a crucial role in the diagnosis of thalassemia, which includes:
• Complete Blood Count (CBC): Decreased hemoglobin levels and microcytosis (low MCV).
• Peripheral Blood Smear: Presence of target cells and abnormal red blood cell morphology.
• Hemoglobin Electrophoresis: Determination of HbA, HbA₂, and HbF levels.
• Genetic Testing: Confirmation of genetic mutations, especially in premarital screening programs.
Treatment and Follow-Up
Treatment depends on disease severity and includes:
• Regular blood transfusions for severe cases
• Iron chelation therapy
• Splenectomy in selected cases
• Bone marrow transplantation as a curative option for some patients
• Regular monitoring of iron levels and cardiac and liver function
Prevention and the Role of Health Awareness
Premarital screening programs and genetic counseling are among the most effective strategies for preventing the spread of thalassemia, particularly in communities with high rates of consanguineous marriages. Health education also plays a key role in improving patients’ quality of life and reducing complications.
Conclusion
Thalassemia is a chronic inherited disease that requires early diagnosis and comprehensive therapeutic intervention. Medical laboratories remain central to detection and follow-up, while prevention and awareness programs represent the cornerstone for reducing the future prevalence of the disease.
Al-Mustaqbal University is the first university in Iraq
Department of Medical Laboratory Techniques – First in the Iraqi National Ranking.