
Scientific article for teaching Prof. Dr. Habeeb sahib Naher entitled: -Sickle cell disease
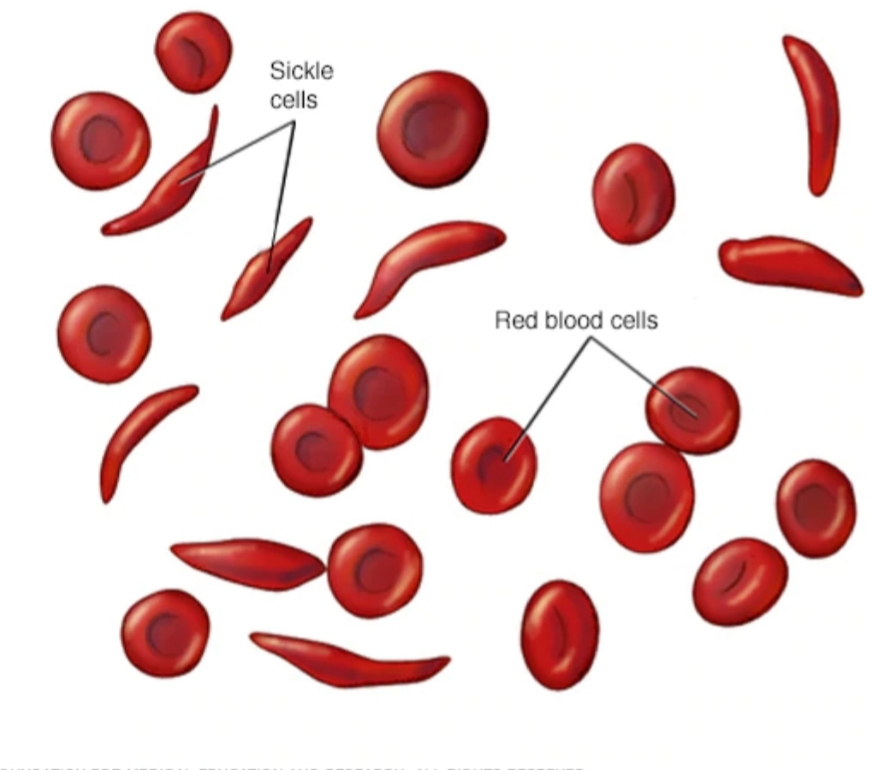
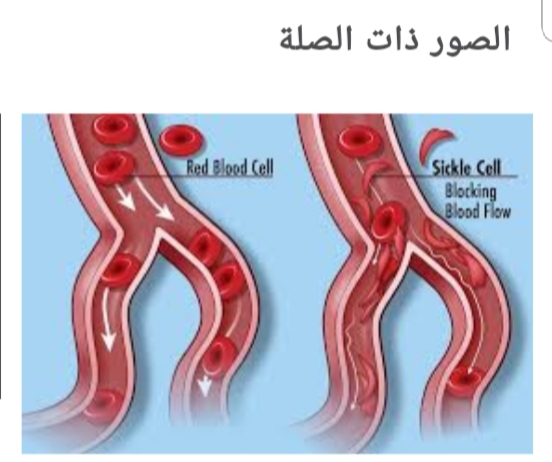
Sickle cell disease<br />By Prof. Dr. Habeeb sahib Naher<br />Sickle cell disease (SCD): It is a group of inherited red blood cells disorder. The most common type is known as sickle cell anemia (SCA). It results in an abnormality in the oxygen-carrying protein (hemoglobin) found in red blood cells. This leads to a rigid, sickle-like shape. Sickle cell disease typically begin around 5 to 6 months of age. <br />Symptoms are long term pain, swelling in hands and feets, anemia and bacterial infections. <br />Sickle cell disease occurs when person inherits two abnormal copies of the haemoglobin gene, one from each parent. This gene occurs in Chromosome 11.<br /> There are several types ot sickle cells depending mutation in each haemoglobin gene. An attack can be set off by temperature changes, stress, and dehydration. A person with a single abnormal copy does not usually have symptoms and is referred to as carrier. Diagnosis is by a blood test and some countries test all babies at birth for the disease. Diagnosis is also possible during pregnancy. The care of people with sickle cell disease may include infection prevention with vaccination and antibiotics, high fluid intake, folic acid supplementation, and pain medication. Other measures may include blood transfusion. Some people can be cured by a transpclant of bone marrow cells. <br />About 80% of sickle cell disease cases are believed to occur in Sub-Saharan Africa. In 2015, it resulted in about 114,800 deaths.<br />* Signs and syptoms. <br />Signs of sickle cell disease usually begin in early childhood. The severity of symptoms can vary from person to person. It may lead to various acute and chronic complications, several of which have a high mortality rate.<br />* Sickle cell crisis.<br />Sickle cell crisis may includ plastic crisis, sequestration crisis and hemolytic crisis. Most episodes of sickle cell crises last between five and seven days.<br />* Vaso-occlusive crisis<br />The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis and often organ damage. The frequency, severity, and duration of these crises vary considerably. Painful crises are treated with hydration, analgesics and blood transfusion.<br />For more severe crises, most patients require inpatient management for intravenous opioids.<br />* Splenic sequestration crisis Because of its narrow vessels and function in clearing defective red blood cells, the spleen is frequently affected. It is usually infarcted before the end of childhood in individuals suffering from sickle cell anemia. This spleen damage increases the risk of infection.<br />Splenic sequestration crises are acute, painful enlargements of the spleen, caused by intrasplenic trapping of red cells and resulting in a precipitous fall in haemoglobin levels with the potential for hypovolemic shock. If it is not treated, patients may die within 1–2 hours due to circulatory failure. Management is supportive, sometimes with blood transfusion. However,these crises are transient; they continue for 3–4 hours and may last for one day.<br />*Acute chest syndrome (ACS)<br />It is the second-most common complication and it accounts for about 25% of deaths in patients with SCD. Most cases present with vaso-occlusive crises, and then develop ACS.<br />* Aplastic crisis: It is acute worsenings of the patient's baseline anaemia, producing pale appearance, fast heart rate and fatigue. This crisis is normally triggered by Parvovirus B19, which directly affects production of red blood cells by invading the red cell precursors and multiplying in and destroying them. Parvovirus infection almost completely prevents red blood cell production for two to three days. <br />* Haemolytic crisis: Hemolytic crisis is acute accelerated drops in haemoglobin level. The red blood cells break down at a faster rate. This is particularly common in patients with coexistent G6PD deficiency. Management is supportive, sometimes with blood transfusions.